
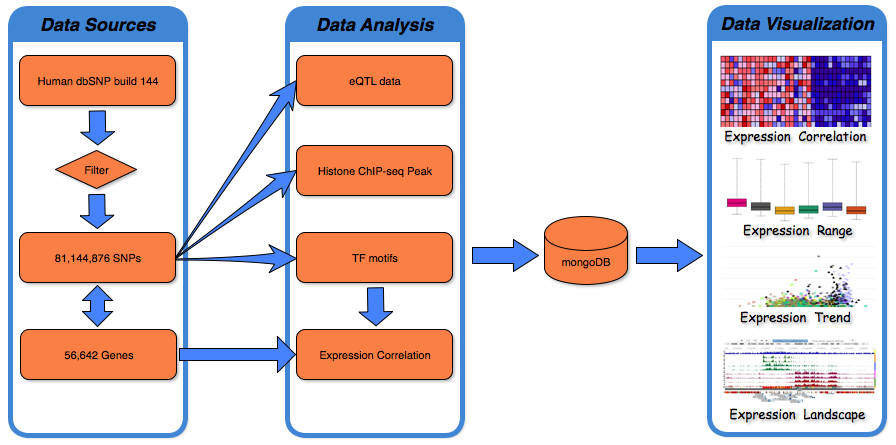
Human dbSNP build 144 was downloaded from ftp.ncbi.nih.gov/snp, which includes 84,435,229 SNPs records, 1,591,294 insertions records, 2,595,517 deletions records, 33,234 indel records and 110 Multiple Nucleotide Polymorphisms (MNPs) records. After filtering redundant records, 81,144,876 of 84,435,229 biallelic SNPs were used to generate functional annotations and were curated by the FeatSNP database. (More details in Data Summary)
The genome coordinates (hg19) of 81,144,876 SNPs were used to associate SNPs to their nearest genes based on 56,642 records of GENOCDE gene annotation Release 19(GRCh37.p13). To predict the impact of allele-specific TF binding affinity by SNPs switching, the Position Weight Matrix (PWM) of 519 vertebrate TFs were collected from JASPAR(Mathelier, et al., 2016). The reference and alternate alleles for each SNP with flanking 10bp of genomic sequences both upstream and downstream were obtained from the UCSC Genome Browser. FIMO1 was used to scan the 21bp sequence to identify binding motifs matching any of the 519 TF PWMs. Only instances where a motif in the sequence:
1,259 transcriptome datasets from 13 brain tissues generated by the GTEx Consortium 3 were used to calculate the Pearson correlation between each SNP associated gene and predicated binding TFs by using bigcor in the R package propagate (v1.0.4). Lowly expressed genes and TFs (mean expression in one tissue less than 0.2RPKM) were removed. The correlation and gene expression in 13 brain tissues were visualized by using JavaScript package Highcharts. eQTL data of 10 brain tissues generated by the GTEx Consortium were negative-log transformed and further visualized by using Highcharts.
Histone modification ChIP-seq data of 10 brain tissues were downloaded from the NIH Roadmap Epigenenomics data portal. Bedtools was used to identify SNPs residing in peaks of 7 histone modification marks (H3K4me3, H3K36me3, H3K27me3, H3K4me1, H3K27ac, H3K9me3, and H3K9ac) that were identified by macs2(Zhang, et al., 2008). To enhance the user experience, the WashU epigenome browser(Zhou, et al., 2015) was embedded in the UI to display the epigenetic landscape in a 400bp region surrounding each SNP. The browser also displays DNA methylation data (Whole Genome Bisulfite Sequencing) of 4 neuronal progenitor and brain tissues generated by the Roadmap Epigenenomics Project, enhanced epilogos visualization (developed by Wouter Meuleman) of all 127 epigenomes, and topologically associating domains (TAD) data of 3 cell lines (GM12878, IMR90, and Hap1)(Rao, et al., 2014; Sanborn, et al., 2015). eQTL data of 10 brain tissues generated by the GTEx Consortium were also visualized on the embedded WashU epigenome browser.
All the functional epigenetic annotations of SNPs were stored as JSON-format documents in the NoSQL database mongoDB4 (v3.2.7).
To show the data more intuitively, data visualization used heatmap (InCHlib5 v1.2.0; Highcharts6 v5.0.2; Clustergrammer7 v1.5.0), boxplot (Highcharts6 v5.0.2), scatterplot (Highcharts6 v5.0.2) and genome browser to explain the relationship between different data.
References: